Direct RNA Sequencing (RNA004) Protocol
Information
This protocol is explicitly for the new SQK-RNA004 Nanopore kit. If you are using the discontinued SQK-RNA002 kit and either an R9.4.1 flow cell or flongle, please see the old Nanopore RNA Sequencing Protocol (SQK-RNA002).
This protocol is a modified version of the Nanopore SQK-RNA004 protocol available here, and the first part series of my m6A Detection Protocol. This protocol will cover the library preparation portion and beginning of the sequencing run, while the m6A detection will be handled in a separate article.
Introduction

This protocol is designed to guide you through the preparation of an RNA library for sequencing through nanopore, adapted from the Oxford Nanopore Technologies Direct RNA Sequencing SQK-RNA004 protocol. Following the sequencing run, you can move onto the analysis protocol for the alignment and detection of RNA modifications using m6ANet or Dorado. At the end of this protocol, I've also included a Troubleshooting section to cover some common issues I've had while sequencing to hopefully streamline your runs. Where possible, I have highlighted deviations from the original protocol in bold red terms to make it easy to identify my changes.
Note that, since this is a modified protocol, you should proceed at your own risk.
Requirements
Below is a list of required reagents and equipment you will need to carry out the protocol. Note that some of these are not necessary for the sequencing run but are highly recommended, such as the Qubit fluorometer, which checks your RNA library's sample quality prior to burning a flow cell. I do not have access to a Qubit, so I will use a total RNA gel and nanodrop results to eyeball RNA quality, but I recommend you use one if you have access to it. Also, additional materials may be required for you to successfully execute this protocol (such as laboratory PPE, pipette tips, pipettes, DNA LoBind tubes, PCR certified H2O, etc., but I have not included them here for simplicity. These items should be standard to your lab and not specific to this protocol.
| Type | Items |
|---|---|
| Materials |
|
| Consumables |
|
| Equipment |
|
| Optional Equipment |
|
Suggested Pre-protocol Preparations
I strongly advise that you remove all reagents (with the exception of enzymes) and leave them out for 10 minutes and allow them to thaw (step 2 in the protocol below). Most of the Oxford Nanopore reagents can sit at room temperature for extended periods of time without worry of degradation. Enzymatic reagents (such as SuperScript III Reverse Transcriptase and T4 DNA Ligase should not be frozen from storage and can be kept on ice. Your RNA sample should be kept on ice, but may need to sit at room temperature for a moment to speed up thawing. I strongly recommend you set a timer for this, so you do not forget about your reagents sitting out if you get distracted during the incubation time.
RNA Library Preparation
WARNING
You need to test your mRNA purity before proceeding. This can be done by saving some of the total RNA and running it on a gel (more information on interpreting those results here) as well as checking the concentration of total and mRNA on a nanodrop. You should have a decent concentration (with mRNA 3-6% of the measured total RNA) with 260/230 and 260/280 ratios of 2.0 or better (mor information on interpreting those results here). A low 260/230 ratio indicates organics in your sample, which can damage the flow cell pores, whereas a low 260/280 ratio indicates proteins, which can clog them. You may want to dilute your input sample so that you're loading your ideal amount in 8 μL of sample, which will also help dilute and contaminants. Higher mRNA amounts than expected (greater than 3-6% of the total RNA) suggests contamination by total RNA or DNA.
Regarding samples that have been frozen: I highly recommend that you pipette your sample 40+ times using an appropriate pipette to draw the full volume of the solution. You should also remeasure the concentration of your sample on the nanodrop, rather than relying on the concentration you obtained prior to freezing. I have had sample library preparations that were sub-par because the concentration dropped following a freeze/thaw.
- Prepare the RNA in nuclease-free water.
- Transfer 330 ng† of poly(A)-tailed RNA or 1 μg of total RNA into a 1.5 mL Eppendorf DNA LoBind tube.
- Adjust† the volume to 8 μL with nuclease-free water.
- Mix thoroughly by flicking the tube to avoid unwanted shearing.
- Spin down briefly in a microfuge.
- †Note: For my best run, I diluted the sample and loaded 8 μL of pure mRNA directly. This was around 333 ng total in 8 μL (~42 ng/μL).
WARNING
Only poly(A)-tailed RNA will eventually be read by the nanopore system. Therefore, if you are interested in non-poly(A)-tailed RNA (such as long non-coding RNA), you will need to poly(A)-tail it separately and purify it prior to the run. Additionally, the old protocol called for 500 ng of poly(A)-tailed RNA, then 50 ng, and now 300 ng (it is unclear at the present on why this was changed). My best run varied from all of these amounts and I get the best results with 330 ng. Finally, according to ONT, there is no difference in using total or mRNA for human samples, however, they acknowledge that this is not the case for other species (such as yeast) and that some users have reported better results with purified mRNA across species. I've found that removing anything that isn't mRNA from the sample provides the best results and slower pore degradation.
- Thaw reagents at room temperature. You can do so by leaving all reagents (except for enzymes) out for 10-15 minutes.
- Ensure the reagents are thoroughly mixed by performing 10 full volume pipette mixes. Do NOT vortex the T4 DNA Ligase.
- Spin down the RT Adapter (RTA), RNA CS (RCS), and RNA Ligation Adapter (RLA), pipette mix and place on ice.
- Thaw the Wash Buffer (WSB) and RNA Elution Buffer (REB) at room temperature and mix by vortexing. Then spin down and place on ice.
- In a 0.2 mL thin-walled PCR tube, mix the reagents in the following order:
WARNING
The NEBNext Quick Ligation Reaction Buffer may have a little precipitate. Allow the mixture to come to room temperature and pipette the buffer up and down several times to break up the precipitate, followed by vortexing the tube for several seconds to ensure the reagent is thoroughly mixed.
| Reagent | Volume |
|---|---|
| RNA Sample | 8† μL |
| NEBNext Quick Ligation Reaction Buffer (see above warning) | 3 μL |
| RNA CS (RCS, Yellow Cap), 110 nM | 0.5 μL |
| RNaseOUT RNase Inhibitor | 1 μL |
| RT Adapter (RTA, Blue Cap) | 1 μL |
| T4 DNA Ligase | 1.5 μL |
| Total | 15 μL |
- †Note: Use 9 μL if low concentration and reduce the water of the next step by 1 μL.
- Mix by pipetting and spin down.
- Incubate the reaction for 10 minutes at room temperature.
- While the reaction incubates, mix the following reagents together in a separate clean 0.2 mL thin-walled PCR tube to make the reverse transcription master mix:
| Reagent | Volume |
|---|---|
| Nuclease-free water | 9† μL |
| 10 mM dNTPs | 2 μL |
| 5x First-strand buffer | 8 μL |
| 0.1 M DTT | 4 μL |
| Total | 23 μL |
- †Note: Use 8 μL if bumped up the RNA amount from step 6 by 1 μL.
- Add the master mix to the 0.2 mL PCR tube containing the RT adapter-ligated RNA from the "RT Adapter ligation" step. Mix by pipetting.
- Add 2 μL of SuperScript III Reverse Transcriptase to the reaction and mix by pipetting.
- Place the tube in a thermal cycler and incubate at 50°C for 50 minutes, then 70°C for 10 minutes, and bring the sample to 4°C before proceeding to the next step.
Note
This can be found on the thermal cycler near Tie's bench as the program RT Nanopore. This is also the point where you can take a break, since this step will take approximately 1 hour and 6 minutes to complete.
- Transfer the sample to a clean 1.5 mL Eppendorf DNA LoBind tube.
- Resuspend the stock of Agencourt RNAClean XP beads by vortexing.
- Add 72 μL of resuspended Agencourt RNAClean XP beads to the reverse transcription reaction and mix by pipetting.
- Incubate on a Hula mixer (rotator mixer) for 5 minutes at room temperature.
- Prepare 150 μL of fresh 70% ethanol in nuclease-free water (i.e., mix 105 μL pure ethanol with 45 μL PCR water in a clean Eppendorf tube).
- Spin down and pellet on a magnet. Keep the tube on the magnet, and pipette off the supernatant (use a p100 if possible to avoid pulling in RNA beads).
Note
For the spin down, I used 2,500g for 1 minute.
- Keep the tube on magnet, and wash the beads with 150 μL of freshly prepared 70% ethanol without disturbing the pellet as described below.
- Keeping the magnetic rack on the benchtop, rotate the bead-containing tube by 180°. Wait for the beads to migrate towards the magnet and form a pellet. Wait 2.5 minutes.
- Rotate the tube 180° again (back to the starting position), and wait for the beads to pellet. Wait 2.5 minutes.
- Remove the 70% ethanol using a pipette and discard.
- Spin down and place the tube back on the magnet until the eluate is clear and colourless. Keep the tubes on the magnet and pipette off any residual ethanol.
Tip
Rotate the tube very fast to avoid the pellet from smearing. You want it to migrate directly across the tube through the ethanol, but otherwise remain intact.
Note
For the spin down, I used 2,500g for 1 minute.
- Remove the tube from the magnetic rack and resuspend pellet (gentle pipetting) in 20 μL nuclease-free water. Incubate for 5 minutes at room temperature.
- Pellet the beads on a magnet until the eluate is clear and colourless.
- Remove and retain 20 μL of eluate into a clean 1.5 mL Eppendorf DNA LoBind tube.
Stop
This is the only stopping point in the protocol. At this stage, the RT-RNA sample can be safely stored at -80°C for later use. If you continue from this step forward, you will need to complete the protocol and load your sample. It will NOT keep after being fully prepared.
- In the same 1.5 mL Eppendorf DNA LoBind tube, mix the reagents in the following order:
| Reagent | Volume |
|---|---|
| RT-RNA Sample | 20 μL |
| NEBNext Quick Ligation Reaction Buffer (see below warning) | 8 μL |
| RNA Ligation Adapter (RLA, Green Cap) | 6 μL |
| Nuclease-free water | 3 μL |
| T4 DNA Ligase | 3 μL |
| Total (including all reagents) | 40 μL |
WARNING
The NEBNext Quick Ligation Reaction Buffer may have a little precipitate. Allow the mixture to come to room temperature and pipette the buffer up and down several times to break up the precipitate, followed by vortexing the tube for several seconds to ensure the reagent is thoroughly mixed.
- Mix by pipetting.
- Incubate the reaction for 10 minutes at room temperature.
- Resuspend the stock of Agencourt RNAClean XP beads by vortexing.
- Add 16 μL of resuspended Agencourt RNAClean XP beads to the reaction and mix by pipetting.
Tip
Check to ensure that the RNA Elution Buffer (REB) is thawing at this point. In my experience, even on ice it tends to remain frozen.
- Incubate on a Hula mixer (rotator mixer) for 5 minutes at room temperature.
- Spin down the sample and pellet on a magnet. Keep the tube on the magnet, and pipette off the supernatant.
Note
For the spin down, I used 3,000g for 1 minute (note the change!).
Tip
Start thawing the Flow Cell Flush Buffer (FCF), RNA Flush Tether (RFT), and Sequencing Buffer (SB) at room temperature in preparation for flow cell priming and loading. If you are loading using the Flongle reagents, also set the Sequencing Buffer, Flongle Flush Buffer, and Loading Beads II out to get to room temperature.
- Add 150 μL of the Wash Buffer (WSB) to the beads. Close the tube lid and resuspend the beads by flicking the tube. Return the tube to the magnetic rack, allow the beads to pellet and pipette off the supernatant.
- Repeat the previous step.
WARNING
Agitating the beads results in a more efficient removal of free adapter, compared to adding the wash buffer and immediately aspirating.
Tip
Make sure the RNA Elution Buffer (REB) is completely thawed at this point.
- Spin down the tube and replace onto the magnetic rack until the beads have pelleted to pipette off any remaining Wash Buffer (WSB).
- Remove the tube from the magnetic rack and resuspend pellet in 13 μL RNA Elution Buffer (REB) by the gently flicking the tube. Incubate for 10 minutes at room temperature.
- Pellet the beads on a magnet for 5 minutes until the eluate is clear and colourless.
- (Optional) Quantify the 1 μL of reverse-transcribed and adapted RNA using the Qubit fluorometer DNA HS assay.
The recovery aim in the final eluate is > 30 ng. Recovery quantities can vary between different inputs and library perparations. However, ONT always recommends taking forward the full volume of RNA library for the best sequencing results. Note that you can view the concentration of your library on the Nanodrop and may or may not get a reading. For a good run, I recorded a slight "hill" peak around 260 nm and around 2.6 ng/μL final concentration. Since your library is double stranded, use the DNA setting on the nanodrop. If you use RNA, you will get an inaccurate reading.
Note that for the loading step, you will want to load the following amounts of RNA for a good sequencing run:
| Flow Cell | Amount of mRNA (in fmol) | Amount in ng (if N50 = 1,400 bases) |
|---|---|---|
| Flongle | 3-20 fmol; 10-15 fmol ideal | ~ 6.751 ng (my best run loaded 2.6 ng/μL in 7 μL, for 18.2 ng total) |
| Standard MinION | 66.86 fmol | 30 ng |
You can also calculate your own loading amounts using the NEBio RNA mass calculator if your RNA is of a different length.
Priming and Loading the SpotON Flow Cell
Once your library is completely prepared, you can now load a flow cell with your sample for sequencing. This section will guide you through this process.
Tip
If this is your first time priming and loading a flow cell, ONT advises that you watch the priming and loading your flow cell video demonstration before your first run.
- Pull out a FLO-MIN004RA flow cell and let it get to room temperature.
- Thaw the Sequencing Buffer (SB), Library Solution (LIS), RNA Flush Tether (RFT) and Flow Cell Flush (FCF) at room temperature. Mix by vortexing and spin down.
- To prepare the flow cell priming mix in a clean 1.5 mL Eppendorf DNA LoBind tube, combine the following reagents:
- Mix by vortexing and spin down at room temperature.
- Open the MinION or GridION device lid and slide the flow cell under the clip. Press down firmly on the flow cell to ensure correct thermal and electrical contact.
- Start your sequencing run on MinKNOW. Before proceeding, you will want to ensure that you can see a large number of pores in the available state, otherwise your flow cell will need to be sent back to ONT.
- Slide the flow cell priming port cover clockwise to open the priming port.
- After opening the priming port, check for a small air bubble under the cover. Draw back a small volume to remove any bubbles:
- Set a P1000 pipette to 200 μL.
- Insert the tip into the priming port.
- Turn the wheel until the dial shows 220-230 μL, to draw back 20-30 μL, or until you can see a small volume of buffer entering the pipette tip.
- Load 800 μL of the priming mix into the flow cell via the priming port, avoiding the introduction of air bubbles. Wait for 5 minutes. During this time, prepare the library for loading by following the steps below.
- In a new 1.5 mL Eppendorf DNA LoBind tube, prepare the library for loading as follows:
- Complete the flow cell priming:
- Gently lift the SpotON sample port cover to make the SpotON sample port accessible.
- Load 200 μL of the priming mix into the flow cell priming port (NOT the SpotON sample port), avoiding the introduction of air bubbles.
- Mix the prepared library gently by pipetting up and down just prior to loading.
- Add 75 μL of the prepared library to the flow cell via the SpotON sample port in a dropwise fashion. Ensure each drop flows into the port before adding the next.
- Gently replace the SpotON sample port cover, making sure the bung enters the SpotON port and close the priming port.
- Place the light shield onto the flow cell, as follows:
- Carefully place the leading edge of the light shield against the clip.
- Gently lower the light shield onto the flow cell. The light shield should sit around the SpotON cover, covering the entire top section of the flow cell.
- Close the device lid.
| Reagent | Volume |
|---|---|
| RNA Flush Tether (RFT, Magenta Cap) | 25 μL |
| Flow Cell Flush (FCF, Clear Cap) | 975 μL |
| Total | 1000 μL |


WARNING
Take care when drawing back buffer from the flow cell. Do not remove more than 20-30 μL, and make sure that the array of pores are covered by buffer at all times. Introducing air bubbles into the array can irreversibly damage the pores. There should always be continuous buffer from the priming port to and across the sensor array.


Tip
If your input RNA sample was of lower concentration than ideal (<300 ng), use the low concentration variation on the right.
| Reagent | Volume (original) | Volume (low conc.) |
|---|---|---|
| Sequencing Buffer (SB, Red Cap) | 37.5 μL | 25 μL |
| Library Solution (LIS, White Cap) | 25.5 μL | 17 μL |
| RNA Libary | 12 μL | 13 μL |
| Total | 75 μL | 55 μL |



WARNING
Install the light shield on your flow cell as soon as library has been loaded for optimal sequencing output. ONT recommends leaving the light shield on the flow cell when library is loaded, including during any washing and reloading steps. The shield can be removed when the library has been removed from the flow cell.


Note
Do NOT force the light shield underneath the clip.

You may now proceed with the sequencing and basecalling step.
Running a Sequencing Run
Starting a sequencing run in MinKNOW should be straight forward. For run settings, I advise that you enable live basecalling using *Dorado* in MinKNOW. This will allow you to keep track of the sample, the quality of the reads you're getting, and the read length histogram. If your sample is of poor quality, you can truncate the run and wash the flow cell. Also keep in mind that you will want to keep pore occupancy at near 100%, since available pores with nothing to sequence will degrade faster (in other words, it is better to slightly over load the flow cell than under load it). For m6A detection, disable Q-score filtering, since lower Q-scores may be generated by high methylation.
Lastly, you can perform a flow cell check prior to starting the sequencing run, but I also advise that you start sequencing before you load any primer or library. Sometimes I've had it where the flow cell check passes (with more than 80% of the pores available), but during actual sequencing, the available pores is much lower (~2%). This is a bad flow cell and should not be used. Starting the sequencing run before loading will allow you to avoid ONT claiming the flow cell was damaged by you during loading.
If your flow cell has a low number of pores available and it has never been used, check to see if it is below warranty. If it is, contact ONT support for a replacement. They will replace flow cells that are below the following thresholds:
| Flow Cell | Minimum number of active pores covered by warranty |
|---|---|
| Flongle Flow Cell | 50 |
| MinION/GridION Flow Cell | 800 |
| PromethION Flow Cell | 5000 |
Troubleshooting
Below is some general information that is helpful during the sequencing run.
- Temperature of the MinION should be stable (a horizontal line). If the temperature of your MinION is fluctuating during the sequencing run, that indicates that your MinION is having difficulty keeping a consistent temperature and may be defective. Check to ensure that the fans on the MinION itself are coming on, otherwise contact ONT for support and/or a replacement.
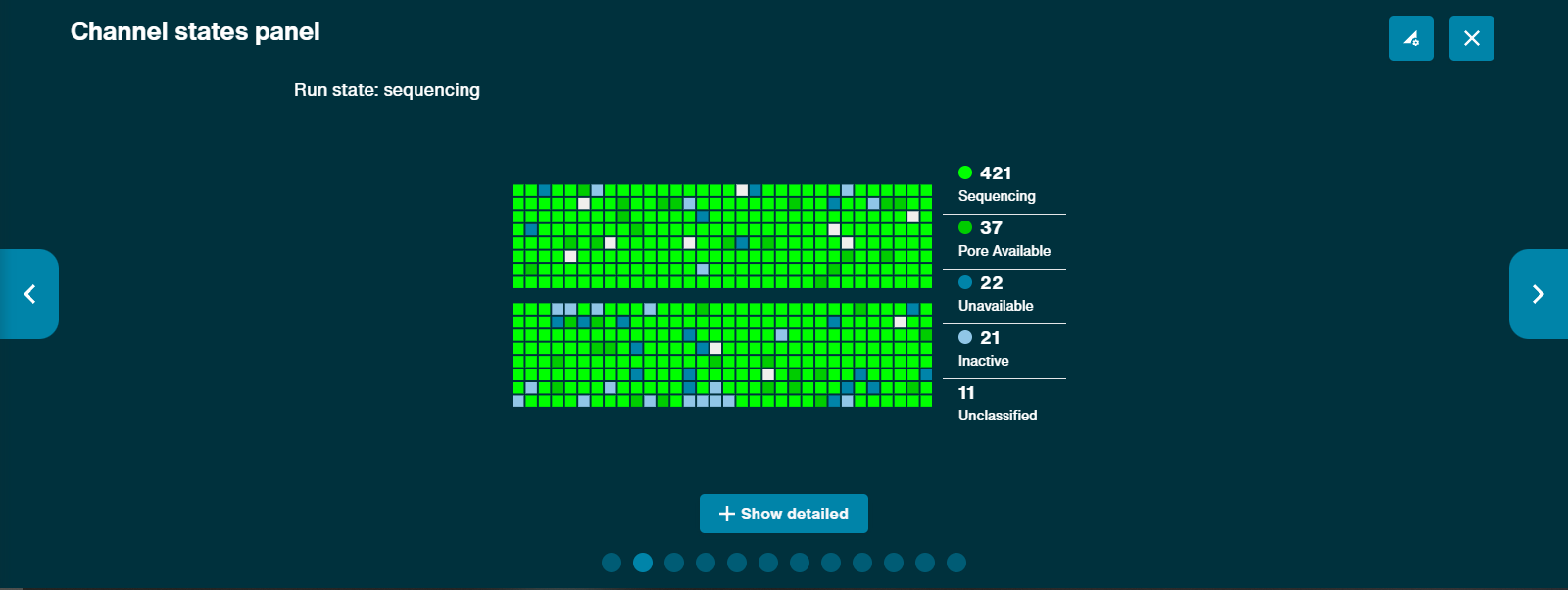
- Large amount of pores in the Inactive state. It has been advised from David Eccles (see this post here) that a large amount of inactive pores in random spots indicates a defective flow cell (example here). Issues with sample preparation and the introduction of air bubbles can cause pores to be inactive, however, this is more likely to occur in distinct regions of the flow cell. It is advised that you start a sequencing run without adding any flush buffer or sample to see how many pores are "available" at the start of your run, then load as necessary. You should see a sea of green pores at the start with no issues (example here). Note: Inactive pores will further separate out into smaller categories, such as "No Pore", when you click "show detailed" at the bottom. It is advisable that you do this if you are troubleshooting a large number of "Inactive" pores.
- Large amount of pores in the Unavailable state. Rapid increases in the number of "unavailable" pores, especially at the start of a library load, can indicate that the pores are becoming clogged. This is especially true if these pores can be recovered by a flow cell wash. Unavailable pores differ from Inactive pores in that unavailable pores are temporarily useless, but may be recoverable, while inactive pores are completely dead source. Note: There are two types of "Unavailable" pores: detailed view and non-detailed view. The non-detailed view ropes in "Active Feedback" pores into this category. So, if you're troubleshooting, make sure you also click "+ show more" on the pore activity page in MinKNOW to see if you're getting a lot of Active Feedback.
- Sequencing RNA will always have a decent number of pores sequencing adapter. Per ONT here: "For RNA in particular, the expanded channels view may show a large proportion of pores sequencing Adapter. This happens because RNA strands are usually shorter than DNA, and the adapter takes up a larger proportion of the strand. Additionally, the RNA sequencing chemistry is optimised for sequencing RNA, whereas the adapter is DNA, and is processed slower. As long as the 'basic' pore activity plot view shows the majority of pores in "Sequencing", a high proportion of Adapter should not be a problem."
- According to Kanhav Khanna, an ONT tech, total RNA and mRNA perform the same for human but not for other species. In my discussions with him, some individuals have reported better results with mRNA (for instance, when studying RNA from yeast), but this hasn't been the case with human RNA. This appears to reflect my runs thus far.
- For the Flongle, aim to load 3-20 fmol of RNA; 10-15 fmol ideally. This can be calculated using the NEBio RNA mass calculator, or the formula on that page. Typically, the average RNA length (N50) is around 1,400 bases, so substitute that into the formula and 10 fmol to determine what amount you should load in ng.
- "Wildflower" Pattern and Flow Cell Quality. Some flow cells appear to have a "wildflower pattern" as it is known on the ONT community forums, since the flow cell has many pores in different states (Active Feedback, Inactive, Sequencing, Adapter, Zero, etc.). You may find a discussion about this on the forums here. As noted by a user, "The good flow cells seem to have a high number of pores at initial check (~1600) whereas flow cells just above the warranty tend to fall off quickly. I saw it with flongles too, those with >80 pores were great, those starting around ~50 died quickly. It seems to be a quality control thing." You should start "sequencing" on your flow cell always to truly check the flow cell's status prior to sample loading.
- Quick Pore Decay can be due to a Number of Factors. Some of these include: large amounts of fragmented RNA/DNA in the sample (you can check this by mapping the control RCS RNA in the Direct RNA Sequencing kit and looking for fragmentation), contaminants present in your sample (detergents, proteins, etc.), poor flow cell quality (flow cells that are old and have been sitting for more than a month, or have low pore availability even if above warranty; see "wildflower" comment above), overloading the flow cell, and/or damaging the pores upon sample loading.
- Unable to draw air bubbles or push solution through the priming port. This is caused by some blockage between the priming port and the waste channel port or crystallization/buildup or residue, and I have seen it happen a few times on a flow cell that was repeatedly run and washed. To remove the blockage there are a few things you can try: 1) opening and reopening the priming port to potentially loosen some blockage (this hasn't really worked for me), 2) add some PCR water to the ports and repeatedly draw a small volume up and down to remove any visible residue, 3) place your finger over waste port 2 and the SpotOn port (with the bung covering the port hole) and gently dispel some air through waste port 1. If it dislodges the block, you should see some fluid move toward the priming port opening. This may require a few tries. 4) As a last resort, the valve itself can be removed by a tiny phillips screwdriver (but I have not tried this method). Additional information on this can be found from threads here and here.
{kind=link}
{kind=link}
| Pore Status | Description |
|---|---|
| Sequencing | Indicates that the pore is currently sequencing. This is the ideal state. Can be either "adapter" sequencing or "strand" sequencing. You can figure out which it is by clicking the "show detailed" box. |
| Pore Available | This means that the pore is available for sequencing, but no strand is currently being sequenced. A large portion of pores in this state usually indicates that you have loaded a small amount of library. |
| Adapter | The pore is sequencing the unligated sequencing adapter only. Reads will initially be classified as adapter until the DNA/RNA strand starts translocating through the pore and MinKNOW is able to reclassify the read. A large portion of adapters sequencing relative to actual sample RNA means that your library preparation was pore. |
| No Pore | No pore was detected in the channel. Part of "Inactive" in non-detailed view. |
| Unavailable | The channel appears to show a pore that is currently unavailable for sequencing. These can be considered "temporarily unavailable" and may be restored with a flow cell wash. Often due to clogging. Part of "Unavailable" (along with "Active Feedback") in non-detailed view. |
| Unclassified | Pore status unknown. |
| Saturated | The channel has switched off due to current levels exceeding hardware limitations. |
| Out of range-high | Current is positive (between 10 and 9999 pA) and unavailable for sequencing. Part of "Inactive" in non-detailed view. |
| Out of range-low | Current is negative (between -5 and -9999 pA) and unavailable for sequencing. Part of "Inactive" in non-detailed view. |
| Zero | Pore currently unavailable for sequencing and the current level is between -5 and 10 pA. According to ONT specialist Kanhav Khanna, this is usually due to osmotic imbalance caused by an air bubble or some detergent in the sample. Look for "swaths" or patches of pores in the Zero state, which may indicate an air bubble in that location. "Inactive" in non-detailed view. |
| Multiple | Multiple pores detected. Unavailable for sequencing. |
| Active Feedback | The channel is reversing the current flow to eject the analyte (DNA/RNA). Part of "Unavailable" in non-detailed view. |
Additional Information
One last thing I wanted to include in this protocol is a list of useful information I have found regarding running mRNA samples through the nanopore. This includes references to the ONT documentation (which may require a login to access the original source) as well as publications. Reference back to it as necessary.
- Poly(A) Tail Required for Sequencing: According to ONT, a poly(A) tail is required for the preparation of the RNA sequencing library. Some RNA molecules, such as long-non-coding RNAs (lncRNAs), rRNAs, tRNAs, and some of your controls (i.e. G. Luciferase and C. Luciferase RNAs) will therefore not be read. It is important, then, to add a poly(A) tail to these molecules enzymatically in vitro prior to the generation of the RNA library using an E. coli poly(A) polymerase (NEB Cat No.: M0276L). Details about this procedure can be found in the ONT Documentation, which also shows a few graphs suggesting optimum incubation time (0.5-1.5 min) at 37ºC (ONT Docs: RNA Polyadenylation).
- Poly(A) Tail Length Bias: ONT has noted in their documentation that there may be a yield bias for mRNAs that have longer poly(A) tails in yeast, but it seems that this may not be the case for humans. Please consult the documentation for more (ONT Docs: Poly(A) Enrichment).
- RNA Contaminants: Various chemicals used in the extraction procedure, including Phenols, Ethanol, Isopropanol, EDTA, NaCl, etc. can affect your RNA. Details of these effects can be found in the ONT Documentation, but the bottom line is that you should avoid contamination during library preparation at all costs (ONT Docs: RNA Contaminants). Additionally, contaminants in your RNA sample may also block the pores in your flow cell, leading to a lot of pores being placed in recovery (blue). NOTE: RNA contaminants, in my experience, and RNA quality greatly affect pore decay. Please see the "RNA Sample Quality and Throughput" point below for more.
- RNA Read Direction 3’->5’: In the RNA sequencing protocol, ONT notes the following: “Please note that, unlike DNA, RNA is translocated through the nanopore in the 3’-5’ direction. However, the basecalling algorithms automatically flip the data, and the reads are displayed 5’-3’.” Useful to keep in mind.
- RNA Stability in Storage: ONT describes in their documentation the stability of RNA over time. If stored properly, RNA should remain relatively stable in the proper buffers at -20ºC for 200+ days (ONT Docs: RNA Stability).
- RNA Stability during Sequencing: RNA will degrade during the sample run over time. Workman et al. found that RNA sequences degrade over the course of a 36 hour run, but this effect was minimal (~5%) in mitochondrial control RNAs over this period, suggesting that a run over the course of 24 hours should exhibit negligible degradation (Workman et al. 2019).
- RNA Sample Quality and Throughput: It is advised that the RNA samples be evaluated for quality via an RNA Integrity Number (RIN) (7+/10) prior to sequencing using an Agilent 2100 Bioanalyzer. This equipment can be pretty pricey, though. Your final RNA library to be sequenced via nanopore should be around 20 ng/uL for a good output. I have found that contaminants and RNA quality greatly affect throughput and pore decay. To avoid this, you should do two things: 1) dilute the input mRNA (before library prep) to around 300 ng / 9 µL and load 9 µL directly. 2) Check the RNA quality via nanodrop and make sure the interpreted results indicate a good RNA sample.
- M6ANet Requires a Minimum of 20 Reads: According to the author here, m6ANet will not generate any inferences for mapped transcripts with fewer than 20 reads. This is a limitation of the software.
- M6ANet Mod Probability: According to the author here, the m6ANet mod probability represents the probability that the modification at that site would be picked up using m6ACE.
- M6ANet Mod Ratio: Again, according to the author here, the m6ANet mod ratio represents the ratio of m6A modifications found to be modified 'using' the m6ANet mod probability. Therefore, mod ratio and mod probability should be positively correlated.
- RNA Read Errors by Base: RNA nanopore reads have higher error rates than for DNA nanopore reads. Additionally, not all basecalling errors are the same. For instance, the nanopore system has a higher error rate confusing C-to-U (3.62%) and U-to-C (2.23%) than G-for-C (0.38%) and C-for-G (0.47%) with Albacore (Workman et al. 2019).
- RNA Read Truncation: In the Workman et al. study, it was also found that there were a number of truncated reads in their mitochondrial poly(A) transcripts, which were used as a control (since they are abundant, vary in length from 349-2,379 nucleotides, and are a single exon). They hypothesize that some possible non-biological causes of this include: 1) the last handful of nucleotides are not read when the pore translocation enzyme detaches the strand near the end, which causes the strand to rapidly move through the pore, 2) ionic current signal artifacts due to enzyme stalls during RNA translocation, or 3) RNA strand breaks (Workman et al. 2019).
- RNA m6A Controls: The Workman et al. study also used an m6A synthetic oligomer (FLuc) as a control for m6A detection. Very useful for you. It is described in the Oligomer Ligation section of the Methods in their paper (Workman et al. 2019).
- Wash Kits do not contain DNase: It appears that the flow cell wash kits do not contain DNase, per some discussions that took place in the comments here. Therefore, it is not likely that your washes will recover flow cell pores following direct RNA sequencing. Perhaps try adding in some RNase?
- Wash Diluent Formula: Someone has claimed that the old wash protocol called for a nuclease flush buffer that consisted of the following: 300 mM KCl, 2 mM CaCl2, 10 mM MgCl2, 15 mM HEPES pH 8.0. Might be useful if you make a homemade buffer for flushing out RNase.
Appendix
- Alignment: A process by which a given DNA or RNA sequence is matched with a reference genome / transcriptome, and therefore given a gene name.
- Bam: A type of DNA or RNA sequencing file containing your reads after they have been mapped to the genome. It is a binary version of the earlier .SAM file, which is where it gets its name.
- Bam.bai: An index file with the same prefix name as a .bam file. This file acts like an external table of contents, which allows algorithms and software to be able to jump to specfic sections of the .bam file without having to read through all the sequences.
- Basecalling: A term used to refer to the process of obtaining direct measurements of ionic current as DNA/RNA is fed through a nanopore, and then using that information to obtain DNA/RNA nucleotide “reads”. Basecalling is achieved by running the raw Fast5 files through the Guppy software.
- Conda: A collection of tools and runtime environment for installing software on a home machine.
- CUDA: Stands for “Compute Unified Device Architecture”, which allows GPU resources to be used for general processing. Developed by and for NVIDIA GPUs.
- Docker: A Linux container whereby you can run Linux code and commands in a virtual environment. I originally used this with an earlier version of this protocol, but it may not be required anymore. Alternative to singularity.
- Fast5: A type of raw file obtained from sequencing DNA or RNA molecules. This can be converted into a Fastq file using the Guppy software provided by Oxford Nanopore.
- Fastq: A type of raw file obtained from sequencing DNA or RNA molecules and houses the sequenced genetic material. Fastq files are often a processed step up from Fast5 files, since they contain some metadata about the sequenced material.
- Flow cell: A term used for the sheet that contains the nanopores that sequence DNA/RNA data, along with their electrodes and sensor chip.
- Guppy: Guppy is a basecalling algorithm provided by Oxford Nanopore Technologies, which reads the fast5 or fastq files obtained from the Oxford Nanopore (such as “MinION”), and generates the DNA/RNA bases.
- MinION / GridION: Two different hardware enclosures that house ONT flow cells while sequencing is occurring and relays that information to a computer.
- Nanopore: Microscopic protein pores embedded in an electrically-resistant polymer membrane and used to process DNA/mRNA strands.
- Nuclease-free Water: Water that has been purified to not be contaminated by non-sample nucleases. This is important for gene sequencing and PCR protocols, to ensure that the material you are working with is only from your sample.
- ONT: A reference to “Oxford Nanopore Technologies”, the manufacturer of the Nanopore we use in the lab. You will see this in a lot of folders that house files from the company, such as “ont-guppy”, which is used to sequence raw nanopore data.
- Python: A type of programming language.
- R: Programming language commonly used by scientists to analyze data.
- Reads: Term used to refer to sequenced genetic material (DNA or RNA).
- Root Folder: The parent folder location that houses most of your files. For the purposes of these guides, we are considering the root folder at
/home/username/(Linux) or/Users/username(Mac). Denoted by “root” or~. - .SAM: A type of file format that holds processed sequencing data. Stands for “Sequence Alignment Map format”.
- Shell Script: A series of code that can be executed at once via the shell (i.e. in Terminal).
- Singularity: A Linux container whereby you can run Linux code and commands in a virtual environment. Alternative to docker.
- Terminal: An application on both Linux and Mac machines that gives you access to the shell whereby you can execute commands.
- XCode: Integrative Development Environment and collection of programming tools provided by Apple, Inc. for developing software.